The book has been out for about 3 weeks and I’ve been thrilled/nervous/pee-my-pants-excited to see my Amazon ranking as well as checking for ratings and reviews on Amazon, Goodreads, and other retail sites like Barnes & Noble, Walmart, Google Books, and Rowman & Littlefield!
For a brief, beautiful, shining moment, it was the #1 New Release in Breast Cancer and Oncology on Amazon, and I have the screenshots to commemorate it!
Pics or it didn’t happen!
Want a sneak peek? Of course you do! Here’s an excerpt from Chapter 16 that deals with an exciting new development in cancer research and treatment – harnessing the patient’s own immune system to seek out and destroy cancer cells through immune checkpoint inhibitors.
EXCERPT
I’ll also take comfort in the fact that we’re getting new weapons in the arsenal for fighting breast cancer. Antitumor immunity is the hottest thing to hit the field of cancer research since the 2001 approval of Gleevec (a game-changer drug used to treat chronic myelogenous leukemia that targets the oncoprotein product of the Philadelphia chromosome that drives the disease) and the 2006 approval of Gardasil (first vaccine targeting the human papilloma virus strains that cause most cervical cancers). Recently Frontiers in Immunology published the history of antitumor immunity efforts leading to the development of immune-checkpoint inhibitors available in the clinic today, the use of engineered T-cells taken from patients and altered to fight their cancer, and oncolytic viruses.2 I’ll go over the basics, including how antitumor immunity works and the challenges we still face in getting tumors to respond.
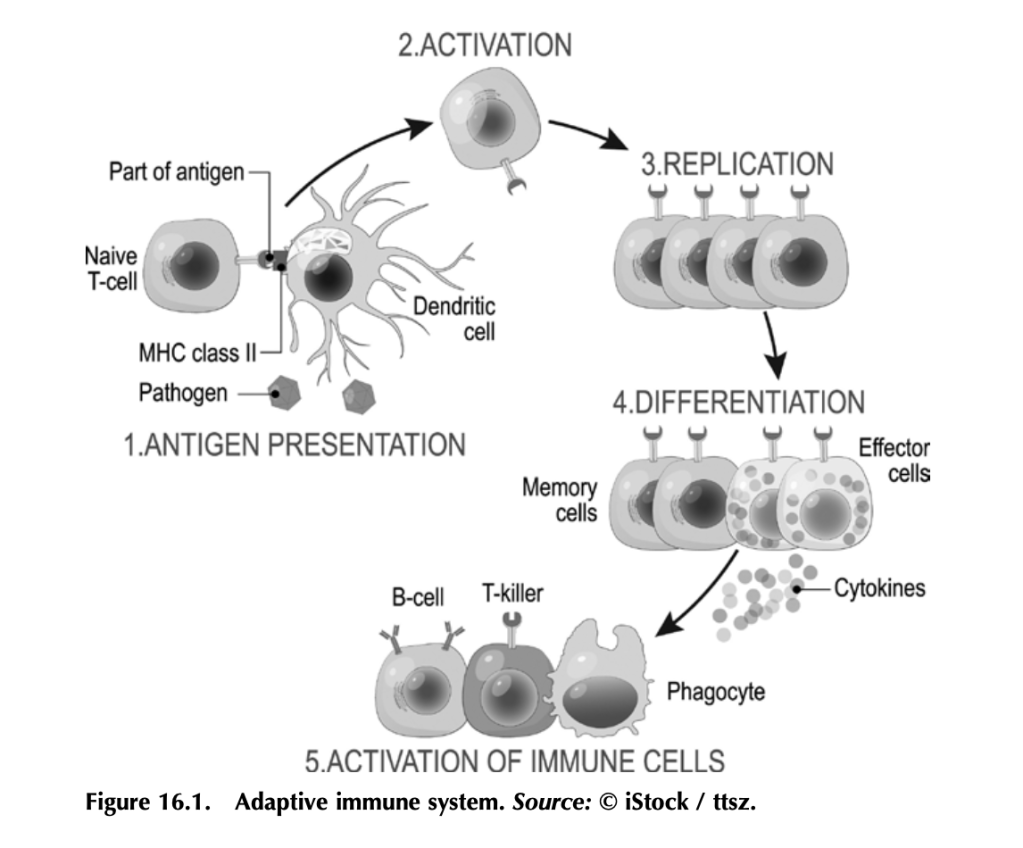
Before we get into how antitumor immunity works, we need to understand how the immune system works to fight infection. It’s a complex beast, but here are some basics. Your immune system functions to mount a rapid and robust defense when your body encounters a pathogen (e.g., a virus or bacteria that causes disease) in your daily life. The arm of the immune system that does this is called the adaptive immune system (figure 16.1). The other arm is the innate immune system, which includes natural barriers like skin, the tiny hairs and mucous in your nose, and stomach acid. The adaptive immune system is what antitumor immunity treatments harness. It is also altered by tumors to suppress tumor immune responses and exploited to work for the tumor. (More on that in a bit.)
The adaptive immune system works like this: Specialized cells identify a potential threat (e.g., an infection), and they carry information about that threat in the form of bits of protein called antigens to other immune cells. If the threat is credible, those immune cells get activated and fight the threat. First the specialized cells that identify a potential threat patrol your body, looking for something suspicious. Cells like macrophages and dendritic cells, which roam around various organs and tissues, find pathogens (a bacteria, virus, or other microbe that causes disease) or unhealthy cells infected by pathogens, and eat them (the fancy term is phagocytosis). Infected or damaged cells send out protein signals called cytokines as a distress call to attract these patrolling macrophages and dendritic cells. While “digesting” the bacteria or infected cell, macrophages and dendritic cells salvage proteins or pieces of proteins—antigens—that identify the bacteria or virus as “other,” and they present these to immune cells, usually in lymph nodes, which in turn mount an immune response. Macrophages and dendritic cells are known as professional antigen presenting cells (APCs).
When activated by APCs, immune cells called B-cells produce antibodies against the antigen, which can do a lot of things to fight an infection. Some antibodies neutralize the pathogen by binding it and stopping it from entering a cell. Other antibodies tag infected cells as a signal for other immune cells to come and kill them. Others coat pathogens or infected cells in a process called opsonization (meaning “the process of making tasty”), which signals other cells like macrophages to come and eat the coated pathogens or cells. Specialized B-cells called memory B-cells store the information about the antigen so your immune system can recognize the pathogen when it hits you again and mount a faster immune response.
Other immune cells called T-cells, which are particularly relevant to antitumor immunity, become activated by APCs and mount a different kind of immune response. Cytotoxic T-cells seek out and kill infected or damaged cells, and helper T-cells help activate B-cells so they make antibodies, activate cytotoxic T-cells, and activate macrophages to go eat nasty invaders and infected cells. Memory T-cells also store information about past infections to mount a rapid, strong response the next time your body sees it.
That’s a simplified but hopefully digestible explanation of immunity and the major players (there are other immune cells, but APCs, B-cells, and T-cells are the biggies).
Memory is key to protection, and memory is built by exposure to pathogens.
Put a pin in that concept for when we get to anticancer vaccines, and also remember what T-cells do for when we get to engineered CAR T-cells and oncolytic viruses.
Working out how to harness your body’s own immune system to fight cancer isn’t a new idea. It’s been under investigation since the nineteenth century. In fact, in chapter 5 we covered the way trastuzumab (trade name Herceptin), a humanized anti-HER2 antibody, targets HER2-expressing breast cancer cells for death. Herceptin and other monoclonal antibodies mimic the natural activity of antibody- producing B-cells to deliver therapies and tag cancer antigen–expressing cells for immune-mediated destruction. But it was the discovery of checkpoint inhibitors—proteins that put T-cells in a state of exhaustion and inactivity in pathways that are exploited by many cancers— that led to the first molecularly targeted therapies designed to boost antitumor immunity. Doctors James Allison and Tasuku Honjo pioneered this Nobel Prize–winning work.3
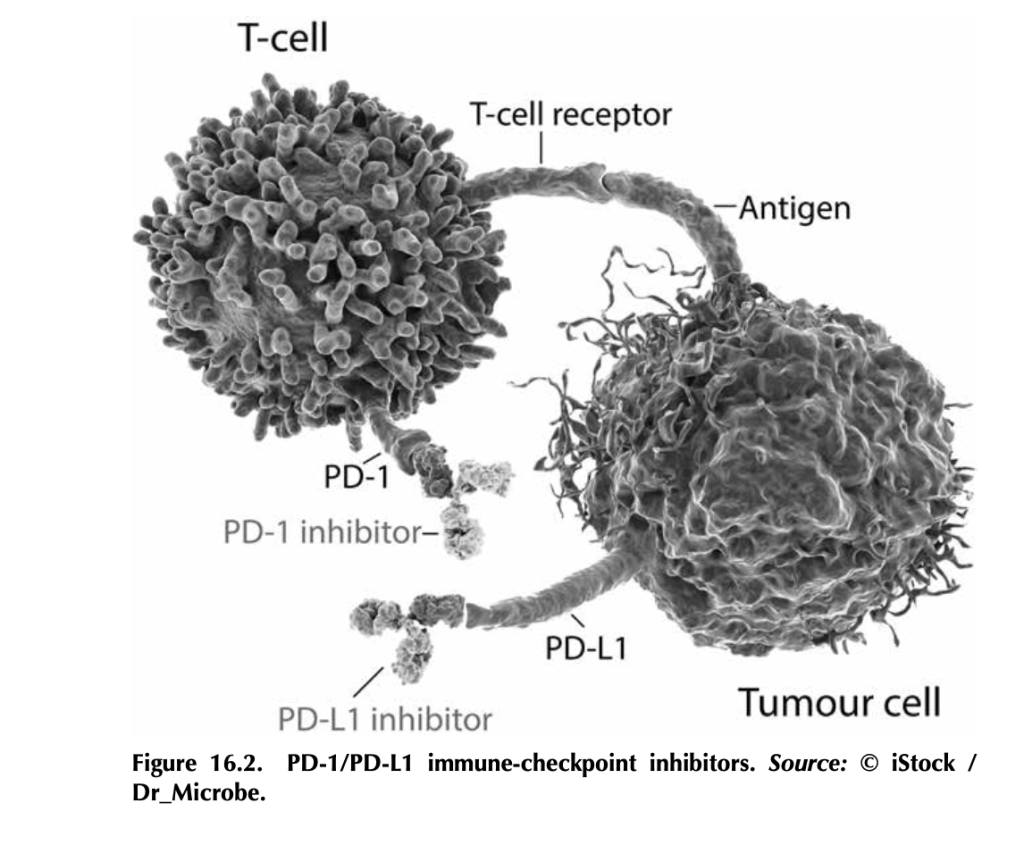
What are immune-checkpoint inhibitors, and how do they work? T- cells, particularly cytotoxic T-cells that actively kill their targets, bind to antigens on tumor cells through their T-cell receptors. But tumor cells, being the adaptable beasts that they are, can produce proteins like PD-L1 (programmed death ligand 1), which bind to PD-1 (programmed cell death protein 1), proteins on T-cells. This interaction tells the T- cell to stand down by tricking it into thinking that the tumor cell is “self” and should be protected. Signaling networks like this normally promote self-tolerance so that your immune system doesn’t attack your own healthy cells (figure 16.2). In tumors, it works by telling tumor- infiltrating T-cells, if present, to go into a state of inactivity. Drugs that target PD-L1—like atezolizumab (trade name Tecentriq), durvalumab (trade name Imfinzi), and avelumab (trade name Bavencio)—and drugs that target PD-1—like nivolumab (trade name Opdivo) and pembrozolimuab (trade name Keytruda)—are FDA-approved mono- clonal-antibody therapies that block interactions between PD-1/PD-L1 to unleash an antitumor immune response.4
Other immune-checkpoint molecules exploited by cancers include cytotoxic T lymphocyte antigen 4 (CTLA-4), the target of the first FDA-approved immune-checkpoint inhibitor ipilimumab (trade name Yervoy). Approved in 2011 for advanced melanoma, this drug had remarkable results. In fact, over 20 percent of the patients enrolled in the initial ipilimumab clinical trials (before the 2011 approval) are still alive and show no evidence of disease (NED).
There’s some incredible potential in targeting checkpoint inhibitors.
CTLA-4 is part of a cellular-signaling pathway that normally fine- tunes immune responses. CTLA-4 and a similar receptor, CD28, are expressed on two different T-cell types: (1) CD4+ helper T-cells, which help activate other immune cells to mediate adaptive immune responses, and (2) CD8+ cytotoxic T-cells, those cells that kill infected cells, damaged cells, and, if properly activated, tumor cells. Antigen- presenting cells make a protein called B7, which can bind to either CD28 or CTLA-4 on T-cells, and the effects on T-cell function are very different depending on what B7 binds. If it binds to CD28, B7 activates T-cell responses as a part of a complex of proteins that includes the T-cell receptor. Binding of B7 to CTLA-4 shuts down T- cell functions. CTLA-4 probably serves as protection from self-antigen recognition by inducing immune suppression, since laboratory mouse models engineered to not express CTLA-4 die from autoimmunity. This is the aspect of CTLA-4 function that gets highjacked by tumor cells. Drugs like ipilimumab block the suppressive activity of CTLA-4, which can allow T-cells to attack tumor cells.5
Here’s the kicker: The tumor actually has to have infiltrating T-cells for this to work, and not all tumors do. Tumors with T-cells that can be activated to fight the tumor are called “hot,” whereas tumors without T-cells are “cold.” One of the most aggressively researched topics in tumor immunology right now is how to make a cold tumor hot and thus responsive to antitumor immune therapies.
This is especially important for breast cancer, since most subtypes produce cold tumors. Right now, immune-checkpoint therapies are only approved for advanced triple-negative breast cancers that make the PD-L1 protein. Not all triple-negative breast cancers make PD-L1. Ongoing research is looking to expand the use of immune therapy in inflammatory breast cancer and the HER2+ subtype.6 Hopefully, with more research, we’ll figure out how to make more tumors responsive to immune therapy by making them hot (full of T-cells) and by discover- ing other immune checkpoints that can be targeted.
3. Heidi Ledford, Holly Else, and Matthew Warren, “Cancer Immunologists Scoop Medicine Nobel Prize,” Nature, October 1, 2018, https://www.nature. com/articles/d41586-018-06751-0.
4. See American Cancer Society medical and editorial content team, “Immunotherapy for Breast Cancer,” Treating Breast Cancer, American Cancer Society, Cancer.org, last revised December 3, 2020, https://www.cancer.org/ cancer/breast-cancer/treatment/immunotherapy.html.
6. Devon Carter, “Does Immunotherapy Treat Breast Cancer?” MD Anderson Center (website), University of Texas, March 26, 2021, https://www .mdanderson.org/cancerwise/does-immunotherapy-treat-breast-cancer.h00 -159385101.html.
I recently had the great pleasure of chatting with a dear friend and fellow breast cancer survivor, Ronei Harden-Moroney. She invited me on her livestream to talk about breast cancer—science, personal experiences, and sharing what it’s like to be in this exclusive club that neither of us signed up for but brought us closer all the same.
As you can see, Ronei is an amazing lady and one tough cookie. I hope hearing her story inspires you and gives you hope. You can find her on Facebook, Twitter, Instagram, LinkedIn, and Goodreads. If you’re looking for an editor or writing coach, seriously check her out!
It occurred to me that while I’ve told you that I’m a cancer researcher, you might not know what that actually means. There are many kinds of researchers who conduct many diverse types of cancer research, as detailed here. All are important and complimentary, and they often overlap. I am an academic (work at a University) laboratory researcher in the broad field of Cell Biology, with a focus on Cancer Biology and Cancer Treatment research, specifically working as a “wet lab” researcher. This means I conduct and supervise hands-on experimental research with cells in a dish, mouse models, and tissue/cell extracts (where we grind up or pulverize tissues and cells, separate them into their components like DNA, RNA, or protein, and analyze them using molecular biology or biochemistry). Other researchers use computational models and datasets to conduct their “dry lab” research.
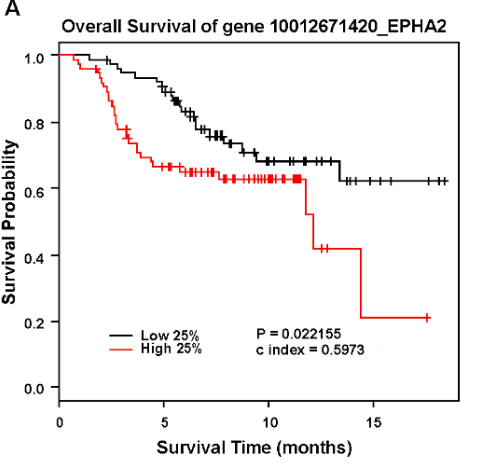
Analysis of EPHA2 expression in human breast cancer patients correlated with poor overall survival. Source here.
Both types of research are important, and one informs and shapes the other. For example, I’ll use information found in large databases generated by dry labs that containing data from actual human cancers (e.g. cBioPortal for Cancer Genomics, Kaplan-Meier plotter, and The Human Cancer Metastasis Database) to find clues about how the gene product molecules I study might be driving cancer cell growth, survival, and invasion. The data I generate then feeds back into these databases, linking known functions in laboratory models along with data about where these gene product molecules are expressed and at what level in human cancers. In fact, all of the cancer research fields listed in the link feed into and fuel each other. Science these days is multi-disciplinary, meaning scientists from diverse fields bring their expertises to the table in order to do better, more advanced, more impactful science. Case in point – I’m working with Dr. Craig Duvall, Biomedical Engineer right now, applying his cutting-edge nanoparticle and carrier technologies to targeting the expression of cancer-driving genes in the cell culture and mouse models in my laboratory.
What is it exactly that you do do? Young Frankenstein, one of the BEST films of all time!
So, what is it exactly that I do…do?
These days, I split my time between the bench (doing actual experiments, which is why I became a scientist in the first place) and the office (doing endless paperwork as quickly and as efficiently as possible so I can get back to the bench). I also supervise a phenomenal medical student and co-mentor insanely smart graduate students, support and collaborate with a team of amazing junior and senior faculty, write grant proposals (more on that below), write up scientific findings into manuscripts for peer review and publication, prepare and deliver scientific talks, maintain compliance (biosafety, environmental safety, radiation safety, responsible care and use of laboratory animals, etc.), make sure the laboratory staff have what they need to perform their research, make sure equipment gets serviced and is operational, attend faculty meetings, scientific seminars, professional development meetings, student thesis committee meetings.
As far as what I research, I use cell culture and mouse models of breast cancer, including metastatic breast cancer, to test new experimental therapeutics.
Experimental Drug (left) getting into tumor cells in 3D culture better than control (right). Source here.
The goal is to discover more specific, effective, less toxic (looking at you, chemo) treatments for breast cancer. I’ll blog more about specific projects later, but what this normally involves is seeing if the new drug makes cancer cells in a petri dish stop growing and/or die, stops cancer cells in a dish from moving and invading, and if a new drug stops tumors in mice from growing or kills them, and, better yet, if the new drugs can actually shrink the tumors. For more information, see the copy of my NIH Biosketch, the mini-resume that we add to every grant application to prove our published expertise, pasted below.
Lung metastases (green) in mouse. The left panel is control and the right is from a mouse lacking ephrin-A1 expression. Source here.
How did I become a cancer researchers? Lots of school and training! I earned a B.A. in Biology from Maryville College in 1995. After graduating, I completed graduate studies at Vanderbilt University, earning a Ph.D. in Cell Biology in 2000. After graduate studies, I completed postdoctoral training in the laboratory of Jin Chen at Vanderbilt University Medical Center from 2000-2003, supported by an American Heart Association Postdoctoral Fellowship Award (I was studying tumor blood vessels, so it was legit!) and a Department of Defense Breast Cancer Research Program Postdoctoral Fellowship award, before being promoted to Research Instructor. I was promoted to Research Assistant Professor in 2006, and during that time I earned a K01 career transition award from the National Institutes of Health/National Cancer Institute. (NIH/NCI – the major funding agency for biomedical cancer research in the united states). This led to my first NIH/NCI independent investigator R01 award in 2011. I was promoted to Assistant Professor of Medicine, Tenure Track, in 2015, and am still at Vanderbilt University Medical Center. I am currently supported by 2 NIH/NCI R01 grants as well as funds from my institution that allow me to generate preliminary data necessary to apply for more grants.
Have you spotted a theme? The theme is “grants” or “awards.” One of the most important jobs I have for my research laboratory is to successfully apply for grants – meaning I write up a proposal about the cool science I want to do, explaining how and why it will benefit patients with breast cancer and move the field forward, and I submit it to the funding agency and compete with a bunch of other super smart, top notch scientists for limited research dollars. These days, money is tough to come by. When I first entered the field as an independent scientist, the top 15% of NCI applications were funded (compared with a funding rate of 25% earlier). These days, it’s at 10%. My colleagues and I literally just missed out on getting a really innovative research proposal funded by 1%! I’m worried how Covid-19 will affect funding over the next 5-10 years, too, as are most of my colleagues. Why is that important? Well, if we want the U.S. to remain on the cutting edge of research and innovation, and if we want to keep discovering new and better ways to detect and treat cancer, we need to invest in science, especially academic science. If you are a cancer survivor, know a survivor, or just want to make the world a better place with less cancer, write your representatives Congress to let them know you want support and full funding of the National Institutes of Health and the National Cancer Institute.
Biographical Sketch
OMB No. 0925-0001 and 0925-0002 (Rev. 10/15 Approved Through 10/31/2018)
NAME: Dana M. Brantley-Sieders
eRA COMMONS USER NAME (credential, e.g., agency login): BRANTLDM
POSITION TITLE: Assistant Professor, Medicine/Rheumatology, Vanderbilt University School of Medicine
EDUCATION/TRAINING (Begin with baccalaureate or other initial professional education, such as nursing, include postdoctoral training and residency training if applicable. Add/delete rows as necessary.)
INSTITUTION AND LOCATION
DEGREE (if applicable)
Completion Date MM/YYYY
FIELD OF STUDY
Maryville College, Maryville, Tennessee
B.A.
05/1995
Biology
Vanderbilt University School of Medicine
Ph.D.
06/2000
Cell Biology
Vanderbilt University Medical Center
Postdoctoral
07/2000
Cancer Biology
A. Personal Statement
I have the expertise, leadership, training, and motivation to successfully carry out the proposed investigation of how EphA2 receptor tyrosine kinase contributes to breast cancer bone metastasis, particularly in terms of tumor-osteoclast interactions that mediate osteolysis in clinically relevant in vivo models that mimic human breast-to-bone metastasis. I have a broad background in cancer research, with specific training and expertise in mouse models of breast cancer and host-tumor interactions (genetically engineered mouse models and orthotopic allograft/xenograft models, including PDX), as well as three-dimensional cell culture and co-culture models, and data mining human tissue microarray and patient datasets to validate clinical relevance of findings in my laboratory model systems. I also have experience testing novel experimental therapeutics in clinically relevant models of breast cancer, including metastatic disease. My research includes analysis of breast cancer cell growth (multiple molecular subtypes), survival, invasion, and host-tumor interactions. Dr. Sterling, Dr. Pellecchia, and I have established a fruitful collaboration that will continue as a part of this exciting investigation
Werfel, T.A., Wang, S., Jackson, M.A., Kavanaugh, T.E., Joly, M.M., Lee, L.H., Hicks, D.J., Sanchez, V., Ericsson, P.G., Kilchrist, K.V., Dimobi, S.C., Sarett, S.M., Brantley-Sieders,D.M., Cook, R.S., and Duvall, CL. (2018) Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res 78:1845-1858. PMID: 29358172. PMCID: PMC5882532.
Sarett, S.M., Werfel, T.A., Lee, L., Jackson, M.A., Kilchrist, K.V., Brantley-Sieders, D., and Duvall, C.L. (2017) Lipophilic siRNA targets albumin in situ and promotes bioavailability, tumor penetration, and carrier-free gene silencing. PNAS 114: E6490-E6497. doi: 10.1073/pnas.1621240114. Epub 2017 Jul 24. PMID: 28739942. PMCID: PMC5558996.
Song, W., Hwang, Y., Youngblood, V.M., Cook, R.S., Balko, J.M., Chen, J., and Brantley-Sieders, D.M. (2017) Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene 36: 5620-30. PMID: 28581527. PMCID: PMC5629103.
Shiuan, E., Inala, A., Wang, S., Song, W., Youngblood, V., Chen, J., and Brantley-Sieders, D.M. (2020). Host deficiency in ephrin-A1 inhibits breast cancer metastasis. [version 2; peer review: 3 approved]. F1000Research 2020, 9:217 (https://doi.org/10.12688/f1000research.22689.2). PMID: 32399207. PMCID: PMC7194498.
B. Positions and Honors
Positions and Employment
Postdoctoral Fellowship, Vanderbilt University School of Medicine
Research Instructor, Vanderbilt University School of Medicine
2006-2015 Research Assistant Professor of Medicine, Vanderbilt University School of Medicine
2015-present Assistant Professor of Medicine, Vanderbilt University School of Medicine
Other Experience and Professional Memberships
1998 Molecular Biology and Pathology of Neoplasia, Edward A. Smuckler Memorial Workshop,Keystone, Colorado
1998-present Member, American Association for Cancer Research
2002 Harvard Medical School Department of Continuing Medical Education and Massachusetts General Hospital Department of Radiation Oncology Seventeenth Annual Offering of Critical Issues in Tumor Microcirculation, Angiogenesis, and Metastasis; Biological Significance and Clinical Relevance Workshop, Cambridge, Massachusetts
2005 National Cancer Institute (NCI)-sponsored Organotypic Models Training Program; received training in orthotopic tumor cell transplantation in mice within several organs, including mammary gland fat pad, bone, lung, spleen, pancreas, bladder, and cecum in the laboratory of Dr. Isaiah J. Fidler, MD Anderson Cancer Center, Houston, Texas
2007-present Ad hoc reviewer for Nature, Cancer Research, PLoS One, Oncogene, Clinical Cancer Research, Neoplasia, European Journal of Cell Biology
2009-2016 Peer reviewer Department of Defense Breast Cancer Research Program
2012 Peer reviewer NCI TME study section
Honors
1997-1998 Department of Defense Breast Cancer Pre-doctoral Fellowship
1998-1999 Dissertation Enhancement Award, Vanderbilt University Graduate School
1998-1999 Coordinator for Developmental Biology Student Organization, Vanderbilt University
2000-2001 Public Health Service Vascular Biology Postdoctoral Fellowship
2001-2003 American Heart Association Postdoctoral Fellowship
2001-2002 American Heart Association Basic Cardiovascular Science Council
2003 NIH NRSA Postdoctoral Fellowship 1 F32 CA101419-01 (award offered, declined due to overlap with 2003 DOD award)
2003-2006 Department of Defense Breast Cancer Research Program Postdoctoral Fellowship DAMD17-03-1-0379
2006-2011 NCI Mentored Career Development Award K01CA117915
C. Contributions to Science
My early publications from graduate studies directly addressed how signaling pathways that regulate normal mammary epithelial morphogenesis (e.g. NF-kappaB transcription factors) can contribute to hyperplasia, a hallmark of neoplastic transformation. These publications provided the first evidence that NF-kappaB transcription factors are expressed and active in normal mammary epithelium during post-pubertal development, and that IkappaBalpha deletion in mammary epithelium, which promotes constitutive activation of NF-kappaB transcriptional activity, promotes pervasive intraductal hyperplasia in vivo. These studies laid the foundation for investigating the role of these transcription factors in breast cancer, and also provided training for me in animal models and mammary fat pad clearing and transplantation techniques that have formed a cornerstone of my independent research program and contributed to numerous collaborations, including those with Dr. Chen. I served as primary author for each of these studies and independently designed experiments, interpreted data, and prepared the manuscripts for publication. Funding from my Department of Defense Breast Cancer Pre-doctoral Fellowship award supported this work. I also contributed directly to collaborations that led to publication of work related to the role of NF-kappaB transcription factors to development and disease as a part of my graduate studies.
Brantley, D.M., Yull, F.E., Muraoka, R.S., Hicks, D.J., Cook, C.M., and Kerr, L.D. (2000) Dynamic expression and activity of NF-kappaB during post-natal mammary gland morphogenesis. Mech Dev 97:149-55. PMID: 11025216.
Brantley, D.M., Chen, C.-L., Muraoka, R.S., Bushdid, P. B., Bradberry, J. L., Kittrell, F., Medina, D., Matrisian, L. M., Kerr, L.D., and Yull, F. E. (2001) Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell 12: 1445-55. PMID: 11359934. PMCID: PMC34596.
Bushdid PB, Brantley DM, Yull FE, Blaeuer GL, Hoffman LH, Niswander L, Kerr LD. (1998) Inhibition of NF-kappaB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature 392: 615-8. PMID: 9560159.
I continued to pursue the connection between signaling pathways that regulate development and contribute to tumorigenesis and progression during my post-doctoral training, providing the first evidence that EphA2 receptor tyrosine kinase regulates angiogenesis and tumor neovascularization. These publications showed that EphA2 regulates endothelial cell assembly and motility through a PI3K/Rac1-GTPase-dependent mechanism and regulates tumor angiogenesis in cooperation with the VEGF signaling pathway in vivo, providing novel insight on the molecular regulation of tumor angiogenesis and host-tumor interactions. I served as primary author for each of these studies and independently designed experiments, interpreted data, and prepared manuscripts for publication. Funding from my American Heart Association and Department of Defense Breast Cancer Postdoctoral Fellowship awards supported this work.
Brantley, D. M., Cheng, N., Thompson, E. J., Lin, Q., Brekken, R. A., Thorpe, P. E., Muraoka, R. S., Pozzi, A., Jackson, D., Lin, C., and Chen, J. (2002). Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene 21: 7011-26. PMID: 12370823.
Brantley-Sieders, D. M., Caughron, J., Hicks, D., Pozzi, A., Ruiz, J. C., and Chen, J. (2004). EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci 117: 2037-49. PMID: 15054110.
Brantley-Sieders, D.M., Fang, W.B., Hicks, D.J., Zhuang, G., Shyr, Y., and Chen, J. (2005) Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J 19: 1884-6. PMID: 16166198.
Brantley-Sieders, D.M., Fang, W.B., Hwang, Y., Hicks, D., and Chen, J. (2006) Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism by EphA2 receptor and Vascular Endothelial Growth Factor (VEGF) in mice. Cancer Res 66: 10315-24. PMID: 17079451.
As PI or co-investigator on several university- and NIH-funded grants, I laid the groundwork for an independent research program by showing that (1) EphA2 receptor tyrosine kinase is necessary for normal mammary epithelial morphogenesis, (2) EphA2 receptor tyrosine kinase promotes mammary tumorigenesis and metastasis in vivo in HER2-dependent models of breast cancer through physical and functional interaction with HER2 and activation of Ras/Erk and RhoA signaling, and, (3) demonstrating clinical relevance of these observations by interrogating patient mRNA datasets and human tissue microarrays to show that high levels of EphA2 correlate negatively with overall and recurrence-free survival in human breast cancer across multiple subtypes.
Brantley-Sieders, D.M., Zhuang, G., Hicks, D., Fang, W.B., Hwang, Y., Cates, J.M.M., Coffman, K., Jackson, D., Bruckheimer, E., Muraoka-Cook, R.S., and Chen, J. (2008) EphA2 receptor tyrosine kinase amplifies ErbB2 signaling, promoting tumorigenesis and metastatic progression of mammary adenocarcinoma. J Clin Invest 118: 64-78. PMID: 18079969. PMCID PMC2129239.
Brantley-Sieders, D.M., Jiang, A., Sarma, K., Badu-Nkansah, A., Walter, D.L., Shyr, Y., and Chen, J. (2011) Eph/ephrin profiling in human breast cancer reveals significant associations between expression level and clinical outcome. PLoS One 6: e24426. PMID: 21935409. PMCID: PMC3174170.
Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, Jackson D, Muraoka-Cook R, Arteaga C, Chen J. (2010) Elevation of receptor tyrosine kinase EphA1 mediates resistance to trastuzumab therapy. Cancer Res 70: 299-308. PMID: 20028874. PMCID: PMC3859619.
My independent research career continues to focus on molecular mechanisms that regulate breast tumorigenesis, host-tumor interactions, and metastatic progression in clinically relevant cell culture and in vivo models. Work initiated in my mentor’s laboratory and supported by an NCI K01 Career Development Award pioneered a role for angiocrine factors regulated by EphA2 in tumor cell growth and invasion in culture and in vivo, providing the first evidence that inhibition of the tumor suppressive angiocrine factor, Slit2, by EphA2 receptor tyrosine kinase promotes tumor cell proliferation and invasion. These studies became the basis of my first independent NIH/NCI R01 grant (CA148934) and publications dissecting the molecular mechanisms through which EphA2 receptor and ephrin-A1 ligand cooperate with VEGF and Slit2 to modulate normal vascular remodeling and tumor angiogenesis in vivo. I served as primary author for the first study and senior author/PI for subsequent studies. I have also recently initiated a collaborative investigation of the role of Rictor/mTORC2 in mammary epithelial morphogenesis and breast cancer with Dr. Rebecca Cook.
Youngblood, V.Y., Wang, S., Song, W., Walter, D., Hwang, Y., Chen, J., and Brantley-Sieders, D.M. (2015)Elevated Slit2 activity impairs VEGF-induced angiogenesis and tumor neovascularization in EphA2-deficient endothelium. Mol Cancer Res. 13:524-37. PMID: 25504371. PMCID: PMC4416411.
Morrison-Joly, M., Hicks, D.J., Jones, B., Sanchez, V., Estrada, M.V., Young, C., Williams, M., Rexer, B.N., Sarbassov, D.D., Muller, W.J., Brantley-Sieders, D., and Cook, R.S. (2016) Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res 76:4752-64. PMID: 27197158.
In addition to the contributions described above, with a team of collaborators, my experience in manipulation of the mouse mammary gland, including xenograft/allograft models, has directly promoted numerous studies elucidating the molecular mechanisms that regulate breast cancer growth/survival, metastatic progression, and host-tumor interactions. Moreover, these studies have benefitted the community at large (e.g. 2012 PLoS One community profiling study provided data for resource allocation requests by Susan G. Komen for the Cure Middle Tennessee Affiliate) and have forged collaborations that will be key in developing new research directions. I served as a collaborator on these studies, contributing to experimental design, interpretation of data, and manuscript preparation/application for funding (some projects).
Takahashi, K., Sumarriva, K., Kim, R., Jiang, R., Brantley-Sieders, D.M., Chen, J., Mernaugh, R.L., and Takahashi, T. (2016) Determination of the CD148-interacting region in thrombospondin-1. PLoS One 11: 5):e0154916. doi: 10.1371/journal.pone.0154916. eCollection 2016. PMID: 27149518. PMCID: PMC4858292.
Young, C.D., Zimmerman, L.J., Hoshino, D., Formisano, L., Hanker, A.B., Gatza, M.L., Morrison, M.M., Moore, P.D., Whitwell, C.A., Dave, B., Stricker, T., Bhola, N.E., Silva, G.O., Patel, P., Brantley-Sieders, D.M., Levin, M., Horiates, M., Palma, N.A., Wang, K., Stephens, P.J., Perou, C.M., Weaver, A.M., O’Shaughnessy, J.A., Chang, J.C., Park, B., Liebler, D.C., Cook, R.S., and Arteaga, C.L. (2015) Activating PIK3CA mutations induce an EGFR/ERK paracrine signaling axis in basal-like breast cancer. Mol Cell Proteomics 14: 1959-76. PMID: 25953087. PMCID: PMC4587316.
Stanford, J.C., Young, C., Hicks, D., Owens, P., Williams, A., Vaught, D.B., Morrison, M.M., Lim, J., Williams, M., Brantley-Sieders, D.M., Balko, J.M., Tonetti, D., Earp, H.S. 3rd, and Cook, R.S. (2014) Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J Clin Invest 124: 4737-52. PMID: 25250573. PMCID: PMCID: PMC4347249.
Brantley-Sieders DM, Fan KH, Deming-Halverson SL, Shyr Y, Cook RS. (2012) Local breast cancer spatial patterning: a tool for community health resource allocation to address local disparities in breast cancer mortality. PLoS One 7:e45238. PMID: 23028869. PMCID: PMC3460936.
Complete List of Published Work in MyBibliography:
NextGen RNAi delivery to breast tumors for selective mTORC2 blockade.
The goal of this study is to optimize advanced nanocarrier technologies for application to targeting the conventionally undruggable cancer driver mTORC2 in breast cancer, including the impact of systemic rictor-targeting RNAi delivery, alone or in combination with chemo and molecularly targeted therapies, on tumor growth/survival, progression, metastasis, and the tumor microenvironment.
Role: Multi-PI with Craig Duvall and Rebecca Cook – no overlap
The Role of EphA2 Receptor Signaling in Host-Tumor Interactions
The goal of this study is to determine if native, membrane tethered ephrin-A1 ligand activates endothelial expressed EphA2 RTK, linking specific domains of the receptor to initiation of endothelial cell migration and neovascularization.
The Role of EphA2 Receptor Signaling in Host-Tumor Interactions
Role: PI
NIH/NCI (Brantley-Sieders) 04/01/2011-03/31/2017
5R01 CA148934
EphA2 receptor in endothelial cell-mediated tumor progression
The goal of this study is to determine how angiocrine factors secreted by tumor endothelilum enhance tumor cell growth and motility, as well as angiogenesis.
Role: PI
NIH/NCI (Chen and Brantley-Sieders) 07/14/2014-05/31/2019
5R01CA177681
Ephrin-A1 in lipogenesis and breast cancer metastatic progression
The goal of this study is to determine how ligand-independent signaling of EphA receptors in the absence of eprhin-A1 promotes HER2-dependent breast tumor progression, metastasis, and lipid metabolism.
Medical students and their mentors in MCN
Medical Scholar conducting breast cancer research in Dr. Dana Brantley-Seider’s lab for Medical Scholars Program
Vanderbilt University Medical Center
Photo: Anne Rayner; VU
This year I have the great fortune of mentoring a talented and dedicated medical student in my lab, Kalin Wilson. Her interest is in oncology, so it’s a great fit for my ongoing and new research directions. She’s working on two projects with similar goals: to identify and characterize new drug combinations and new experimental therapeutics for triple negative breast cancer in pre-clinical studies. This is an urgent unmet need in the clinic. Triple negative disease is a subtype of breast cancers that do not express hormone receptors (estrogen receptor and progesterone receptor) or cell surface HER2 (amplified in ~25% of breast cancers). These receptors are druggable targets, and their absence limits treatment options for patients with triple negative breast cancer to surgery, radiation, and chemotherapy. Triple negative breast cancers are aggressive and disproportionately affect young women and women of African descent. Our goal is to find molecular targets for new drugs to give women with this type of breast cancer more and better options.
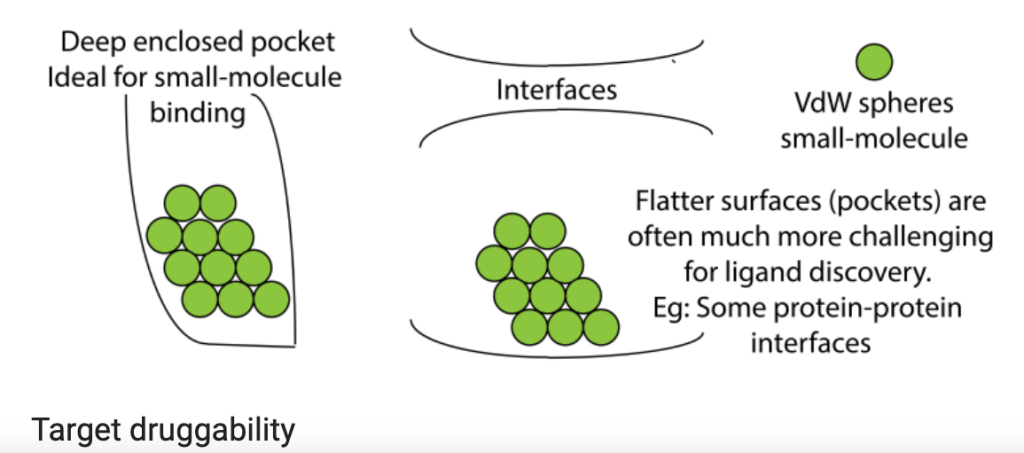
My student’s primary project is to test nanoparticle delivery systems to transport gene therapy to tumors. Many of the genes that drive cancer code for proteins that aren’t easily druggable by small molecules that fit neatly into a unique structural region in the target protein to block its function (e.g. deep enclosed pocket versus flat, relatively uniform interface or surface, as shown in the figure above). But what if we could stop production of cancer-driving proteins at the level of gene expression? This is actually possible in the laboratory setting in a process that exploits messenger RNA, the protein-making instructions that are copied from DNA and used by protein producing cellular machinery (see figure below). The use of small interfering RNA (siRNA) gene therapy, which causes the messenger RNA that encodes the protein’s blueprint to be destroyed, can theoretically stop production of any protein, which would make any target druggable. One of the challenges, however, is delivery of siRNAs to tumors. siRNAs tend to be unstable, so they can be easily destroyed by immune cells or taken up by the liver or kidneys as a part of their normal clearance functions. To overcome those delivery barriers, many biomedical engineers are applying nanotechnology, designing nanoparticles that surround the siRNA molecules. These nanoparticles shield and protect the siRNAs in circulation and can be modified to help homing to the tumor. In collaboration with Dr. Craig Duvall, we are testing nanoparticles delivering siRNA to destroy the blueprint for Rictor, a protein that we believe is essential for tumor cells to grow and prevents them from dying when they’re supposed to. Results so far are promising!
What I hope to give Kalin is a research experience that feeds her passion for science and drug discovery, to foster her natural skills and curiosity, and to keep striving for the goal of bench-to-bedside translational research. What she’s given me is her clinical perspective, something that has enriched my research and inspired me to do more directly translational research with the goal of clinical application. She’s also given me the gift of fearlessness and enthusiasm, which young scientists always possess in abundance and, fortunately, is contagious. The rewards of mentoring the next generation of scientists are many, but the synergy between experience (mentor) and fresh ideas and perspectives (mentee) is perhaps the most valuable.
This is the first in a series of posts dedicated to the science of breast cancer, so let’s start from the beginning with normal breast. In order to understand how cancer forms and grows, you first have to understand how non-cancerous cells behave and function. Why? Because fundamentally, cancer is an aberration of normal function. Cancer cells were once normal cells. A series of events that involve mutation in the cell’s DNA, the genetic blueprint that encodes instructions and specific modifications for that cell’s function that lead to changes in (1) the cell’s ability to divide, (2) the cell’s response to normal programmed cell death, (3) the cell’s ability to repair damaged DNA. These events reprogram the cell’s function and cause uncontrolled, abnormal cell growth, and these changes are alterations in the normal cell programs that maintain the balance between new cell growth and old cell death that maintain healthy cell function.
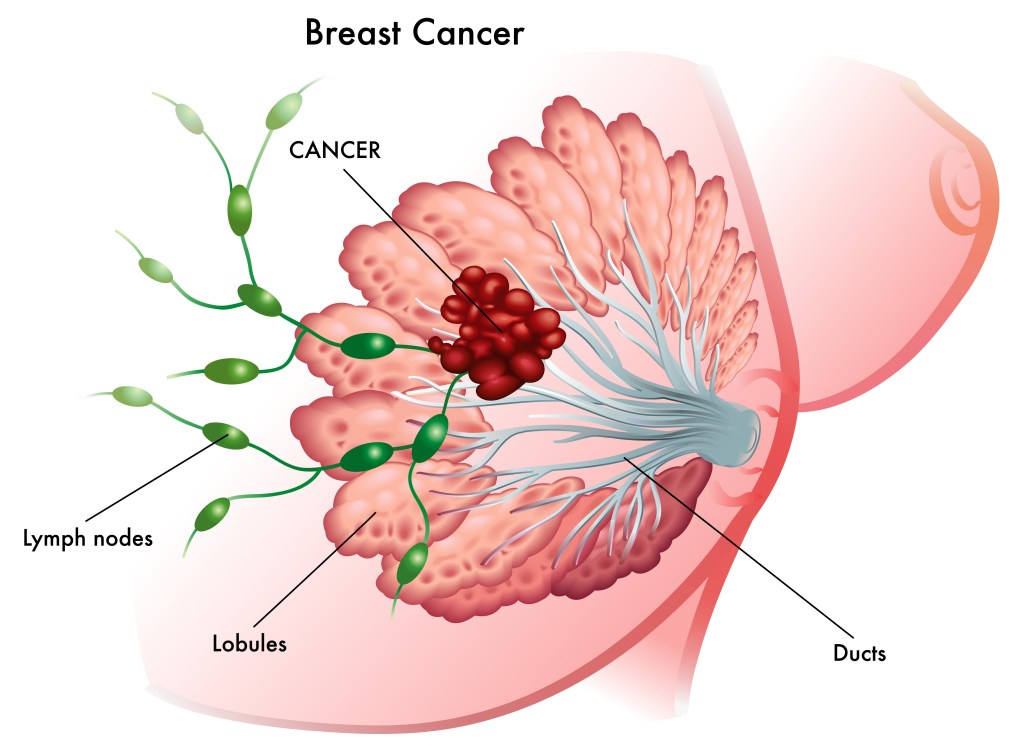
Let’s start with anatomy. Breasts are made up of milk-making (lobules) and shuttling (ducts) glandular epithelial cells anchored by connective tissue and support cells in a sea of squishy fat (adipose tissue). The glandular epithelium goes through a massive growth cycle during pregnancy and becomes a milk factory for nursing young. When the young stop nursing, the factory shuts down, most of the cells die, and the epithelium rests until the next pregnancy. The same cellular programs that control growth and death in these normal cycles become highjacked when a cell begins to transform from normal to cancer. This includes programs regulated by hormones like estrogen and progesterone, as well as cell surface growth factor receptors like HER2, which we will cover in future posts.
Most breast cancers form from ductal epithelial cells, but can form from lobular or other types of cells*. The most important take home message is that breast cancer isn’t a single disease. It is a collection of diseases classified by pathology (how it looks under the microscope) and molecular genetics (which collection of mutations in specific genes contribute to its formation and progression). There are at least five broad subtypes of breast cancer that can be further divided into additional subtypes: (1) Luminal A, which tend to be estrogen and progesterone hormone receptor positive (ER+/PR+) and lack HER2 alteration; (2) Luminal B, which tend to be estrogen receptor positive and can be HER2 positive or negative; (3) HER2-enriched, which tend to be negative for hormone receptors (ER-/PR-) and display amplification (more copies) of the gene encoding HER2 cell surface receptor; and (3) Triple negative, which lack hormone receptors (ER-/PR-) and HER2 amplification (HER2-)**. I’ll cover each of these subtypes in future posts, including the latest research on how they form at the molecular, genetic, and cell biologic level, and current/emerging treatment options.
To wrap things up, I’d like to share with you some of the work I did as a graduate student***, which involved understanding molecular regulation of normal breast epithelial development during puberty. Again, understanding how normal breast epithelium grows and forms as breasts develop is an important first step in understanding how things go wrong in breast cancer. The pictures in (A) show whole-mount preparations of mammary gland (a fancy term for squishing and flattening a small piece of tissue on a slide and staining it to show the epithelium in the sea of fat) of breast epithelium growing to fill the fat of developing breast during puberty. The specialized bulb-like structure (arrow) is called a terminal end bud (TEB). The schematic in (B) shows the structure of cells within the TEB as they grow from the TEB tip out and differentiate into their normal, mature structures in the area behind the TEB. Luminal epithelial cells line the ducts, while myoepithelial cells that surround the lumina structure contain contractile proteins that, like muscle, will eventually squeeze and contract to help milk travel to the nipple. Cap and body cells turn into these cell types when growth stops.