It’s Day 16 of National Breast Cancer Awareness Month! Today’s topic will cover one of the oldest targeted breast cancer drugs developed that is still used in the clinic today—tamoxifen. I’ve been taking it for 3 1/2 years, and millions of other breast cancer survivors with ER+ breast cancer have taken this drug as part of their treatments to prevent recurrence. But how was it discovered? How does it work?
Like many scientific discoveries, the discovery of tamoxifen was an accident. ICI46,474, later named tamoxifen, was first synthesized in 1966 by scientists working for a company that would become AstraZeneca. The goal of the project was to find a new chemical compound that could be made into a birth control drug. Laboratory studies were promising, but they found it didn’t work as a form of contraception in humans. This could have been the end of the story for tamoxifen, but one of the members of the team thought it might work as a breast cancer drug. In 1971, tamoxifen was tested in a clinical trial conducted in the UK on “late or recurrent carcinoma of the breast.” Thankfully, it worked!
How does it work? Since it started out as a candidate contraceptive, it makes sense that it was designed to block estrogen, a female hormone that helps prepare the uterus and uterine lining for pregnancy. It is in a class of drugs known as Selective Estrogen Receptor Modulators (SERMs), which are compounds that compete with the hormone estrogen for binding to its receptor. Normally, when estrogen binds to its receptor in the body, it triggers processes in the cell that make it divide, or produce more cells. This is called proliferation. In cancers with too many estrogen receptors (ER), estrogen in the body makes these cells grow uncontrollably. By binding to estrogen receptors in breast cancer cells, tamoxifen blocks this action and stops breast cancer cells from growing.
Around 70-80% of breast cancers are ER+, meaning that abnormal estrogen receptor activation is a key driver for growth of the breast cancer cells. Tamoxifen was a game changer for women with ER+ disease, reducing the annual breast cancer death rate by 31%. There are other drugs on the market that also block the activity of estrogen or downstream molecules in the estrogen receptor pathway, but tamoxifen remains standard of care for many cases of ER+ breast cancer.
As with any medication, tamoxifen comes with side effects that include: hot flashes, vaginal discharge, nausea, mood swings, fatigue, depression, hair thinning, constipation, loss of libido, dry skin. I experienced hot flashes, vaginal dryness and libido issues, and hair thinning, but they were not as severe as those I experienced with other estrogen blockers (aromatase inhibitors). For me, tamoxifen is a better balance between protection from recurrence and quality of life, but everyone’s physiology and experiences are different.
Be sure to talk to your healthcare providers about any side effects you experience. You don’t have to suffer in silence, and there are options to reduce side effects and improve your quality of life.
It’s the fifth day of National Breast Cancer Awareness Month 2022! Quick update from yesterday – my (hopefully) last breast reconstruction procedure went great! I’m home recuperating and enjoying love from my fur babies and my family. I haven’t looked at the result yet, but the left side of my surgical bra looks fuller. Hooray!
Now, back to the subject of breast cancer molecular subtypes! To recap, breast cancer isn’t a single disease. It is a collection of diseases that cause cells in the breast—specifically the cells that produce and deliver milk to nursing infants called epithelial cells—to grow uncontrollably, forming a tumor. Each breast cancer case is as unique as each person, but they can be classified based on similarities in how they look under a microscope (histology) and on the characteristics of their DNA (molecular).
Molecular breast cancer subtypes, which are crucial diagnostic tools used to determine the best and most appropriate course of treatment, include four subtypes recognized by scientists and clinicians based on their expression of hormone receptors (HR) for estrogen and progesterone (ER and PR) and their expression of the cell-surface receptor HER2: Luminal A, Luminal B, HER2-positive, and Triple Negative Breast Cancer TNBC.
ER = estrogen receptor, PR = progesterone receptor, HER2 = positive for the cell surface receptor HER2, Ki-67 = a marker for how fast cancer cells grow, -ve = negative, +ve = positive. Image Credit Here.
Today’s post is all about the triple negative subtype, which do not express hormone receptors for estrogen and progesterone and also do not express express a cell surface protein receptor called HER2. That’s how it got its name, triple negative, because of the three receptors it does not express. These breast cancers have other ways of growing abnormally that don’t involve hormones or HER2, and they tend to grow very fast. For example, some TNBCs have higher higher than normal levels of EGFR receptors, which are in the same cell surface receptor family as HER2. Like HER2, EGFR normally tell breast cells to grow during normal development in puberty. In cancer, these receptors stay active and make breast cells grow when they shouldn’t, which is a key characteristic of breast cancer. Aside from targeting EGFR, a strategy being tested in clinical trials, people with TNBC have fewer treatment options with targeted therapies (therapies that inhibit hormone receptors and HER2) than other subtypes.
Aside from EGFR receptors, what make TNBC cells grow? There are several molecular pathways that become altered in TNBC. These pathways often function in normal breast epithelial cells telling them to grow when appropriate (cell surface receptors like EGFR, FGFR, and CSF1-R). When receptors on the surface of the cell becomes activated, they send signals to the breast cell that tells it to grow, like when your breasts are growing during puberty. Normally, after puberty, the receptor and related receptors are no longer activated and your breast cells stop growing. In breast cancer, your breast cells make too many receptors, which become constantly activated, making your breast cells grow abnormally, which is one hallmark of cancer. If other changes occur in your breast cells to form a cancerous growth, these receptors make the cancer cells grow uncontrollably.
One interesting characteristic of TNBC is that this subtype often has more interactions with the body’s immune system, meaning that immune cells travel to the tumor, get inside of it, and try to kill cancer cells and eliminate them from the body. Many cancer cells develop defenses against the immune system, using cell surface proteins like PD-1, PD-L1, and CTLA4 to shut down the immune cells. Tumors with lots of immune cells, called immunologically “hot,” may respond well to immune therapies that block the activity of PD-1, PD-L1, and CTLA4. The drug Pembrolizumab, which targets PD-1, is used to treat advanced TNBC. The drug Sacitiuzumab Govitecin was recently approved to treat TNBC. More on that below!
TNBC breast cancer is not as common as HR+ breast cancer, accounting for 15-20% of breast cancers. However, these breast cancers are often more aggressive and faster growing than HR+ breast cancer. They also disproportionately affect younger women and women of African descent. They are diagnosed by a pathologist based on analysis of gene and protein expression if ER, PR, and HER2 present in cancer cells in a biopsy and in the tumor after surgical removal. Low or no expression of these three receptors leads to a diagnosis of TNBC. This type of breast cancer, like most breast cancers, is first treated by surgery to remove the tumor.
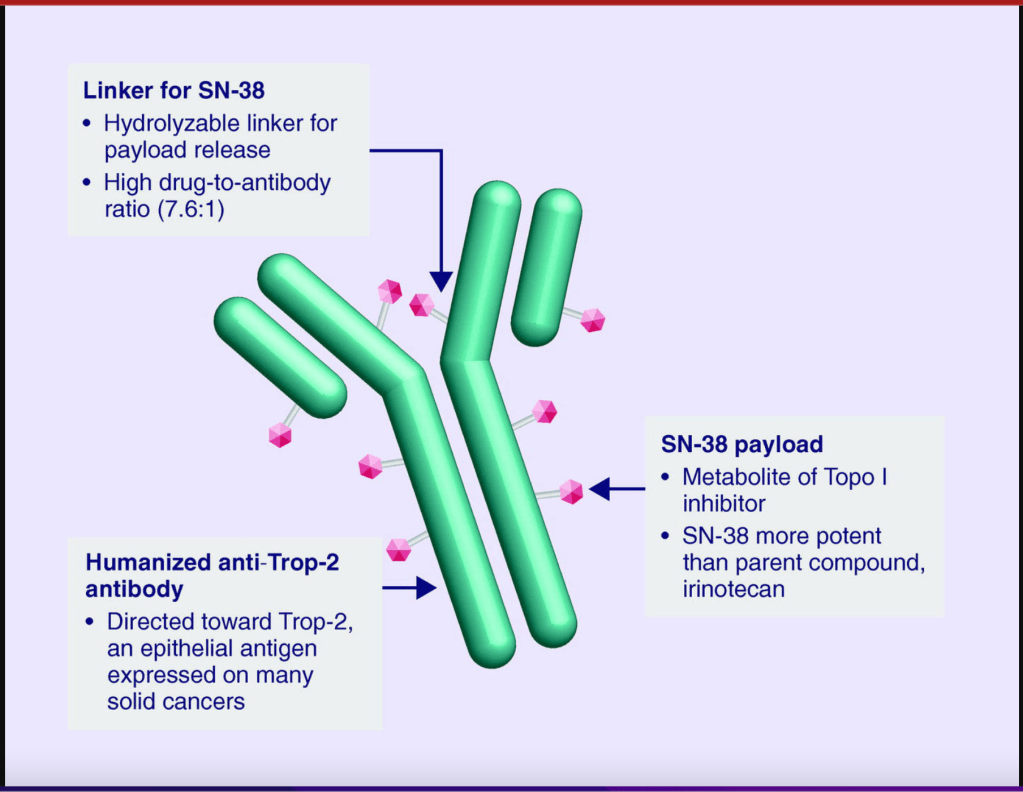
Follow-up treatments include chemotherapy, which was the only option for people diagnosed with TNBC for many decades. Recently, drugs developed specifically to treat TNBC have been approved and are being used in the clinic and undergoing additional testing in clinical trials to refine and optimize their ability to work with other drugs to kill more cancer cells. I discussed Pembrolizumab above. Let’s go over a little bit about Sacituzumab Govitecan. This drug binds to the cell surface protein Trop-2. Many TNBCs have higher than normal levels of Trop-2 on the surface of their cells. Sacituzumab Govitecan, approved in April of 2022, as a treatment for metastatic TNBC, is an antibody-drug conjugate that uses the an antibody against Trop-2 to carry a toxic drug directly to breast cancer cells with high levels ofTrop-2, targeting tumor cells and reducing damage to normal cells and tissues.
These treatments reduce the risk of the cancer from coming back, or recurring. They do come with some not-so-great side effects, which your oncologist should consider and help you with. Quality of life should always be a consideration when it comes to cancer treatment.
For more on TNBC, check out the American Cancer Association. As with other subtypes of breast cancer, early detection increases your chance of survival, so keep up with your routine mammograms and self-exams. Though TNBC is trickier to detect, screening mammography remains a vital tool for early detection of TNBC and other breast cancer cell types.
It’s the third day of National Breast Cancer Awareness Month 2022! To recap, breast cancer isn’t a single disease. It is a collection of diseases that cause cells in the breast—specifically the cells that produce and deliver milk to nursing infants called epithelial cells—to grow uncontrollably, forming a tumor. Each breast cancer case is as unique as each person, but they can be classified based on similarities in how they look under a microscope (histology) and on the characteristics of their DNA (molecular).
Molecular breast cancer subtypes, which are crucial diagnostic tools used to determine the best and most appropriate course of treatment, include four subtypes recognized by scientists and clinicians based on their expression of hormone receptors (HR) for estrogen and progesterone (ER and PR) and their expression of the cell-surface receptor HER2: Luminal A, Luminal B, HER2-positive, and Triple Negative Breast Cancer.
ER = estrogen receptor, PR = progesterone receptor, HER2 = positive for the cell surface receptor HER2, Ki-67 = a marker for how fast cancer cells grow, -ve = negative, +ve = positive. Image Credit Here.
Today’s post is all about the HER2-enriched (also called HER2+) subtypes, which express a cell surface protein receptor called HER2. These breast cancers have higher than normal levels of HER2 receptors, which normally tell breast cells to grow during normal development in puberty. In cancer, these receptors stay active and make breast cells grow when they shouldn’t, which is a key characteristic of breast cancer. Luminal B breast cancers have too much of the cell surface receptor HER2 in addition to having too much estrogen and progesterone receptors (ER/PR +ve), as do breast cancers that do not also express hormone receptors (HER2-enriched).
How does HER2 receptors make breast cancer cells grow? When the receptor on the surface of the cell becomes activated, it sends a signal to the cell that tells it to grow, like when your breasts are growing during puberty. Normally, after puberty, the receptor and related receptors are no longer activated and your breast cells stop growing. In breast cancer, your breast cells make too many HER2 receptors, becoming constantly activated ,making your breast cells grow abnormally, which is one hallmark of cancer. If other changes occur in your breast cells to form a cancerous growth, these HER2 receptors make the cancer cells grow uncontrollably.
HER2+ breast cancer is not as common as HR+ breast cancer, accounting for 10-20% of breast cancers. However, these breast cancers are often more aggressive and faster growing than HR+ breast cancer. They are diagnosed by a pathologist based on analysis of HER2 gene expression and HER2 proteins present in cancer cells in a biopsy and in the tumor after surgical removal. This type of breast cancer, like most breast cancers, is first treated by surgery to remove the tumor. .
Follow-up treatments include chemotherapy andHER2-targeted therapies that block the activity of HER2 on breast cancer cells, including antibody drugs that bind to HER2 receptors and cause them to be degraded by the cell as well as triggering the body’s natural immune system to attack HER2+ tumor cells. These drugs include Trastuzumab and Pertuzumab. A derivative of Trastuzumab called Ado-trastuzumab emtansine (also called T-DM1) is an antibody-drug conjugate that uses the Trastuzumab antibody to carry emtansine chemotherapy directly to breast cancer cells with high levels of HER2, targeting tumor cells and reducing damage to normal cells and tissues.
These treatments reduce the risk of the cancer from coming back, or recurring. They do come with some not-so-great side effects, which your oncologist should consider and help you with. Quality of life should always be a consideration when it comes to cancer treatment.
For more on HER2+ positive breast cancer, check out the American Cancer Association. As with other subtypes of breast cancer, early detection increases your chance of survival, so keep up with your routine mammograms and self-exams.
It’s the second day of National Breast Cancer Awareness Month 2022! Did you know that breast cancer isn’t a single disease? It is a collection of diseases that cause cells in the breast—specifically the cells that produce and deliver milk to nursing infants called epithelial cells—to grow uncontrollably, forming a tumor. Each breast cancer case is as unique as each person, but they can be classified based on similarities in how they look under a microscope (histology) and on the characteristics of their DNA (molecular).
This post and upcoming posts will focus on molecular breast cancer subtypes, which are crucial diagnostic tools used to determine the best and most appropriate course of treatment. There are currently four molecular breast cancer subtypes recognized by scientists and clinicians based on their expression of hormone receptors (HR) for estrogen and progesterone (ER and PR) and their expression of the cell-surface receptor HER2: Luminal A, Luminal B, HER2-positive, and Triple Negative Breast Cancer.
ER = estrogen receptor, PR = progesterone receptor, HER2 = positive for the cell surface receptor HER2, Ki-67 = a marker for how fast cancer cells grow, -ve = negative, +ve = positive. Image Credit Here.
Today’s post is all about Luminal A and Luminal B subtypes, which are hormone receptor-dependent (hormone receptor-positive, also known as estrogen receptor-positive or estrogen receptor/progesterone receptor-positive). These breast cancers have higher than normal levels of receptors for estrogen (ER+) and progesterone (PR+), which normally tell breast cells to grow during pregnancy as they get ready to start producing milk. In cancer, these receptors stay active and make breast cells grow when they shouldn’t, which is a key characteristic of breast cancer. Luminal B breast cancers also have too much of the cell surface receptor HER2, which also makes breast cells grow uncontrollably, contributing to cancer. HER2 positive breast cancer will be covered in the next post.
How do estrogen and progesterone receptors make cancer cells grow? Estrogen produced by your body binds to molecules called receptors. When estrogen or progesterone enters a breast cell, it binds to a partner, called a receptor. When the receptor binds to the hormone, it sends a signal to the cell that tells it to grow, like when you’re pregnant and your breasts are getting ready to make milk for when the baby is born. Normally, after pregnancy and lactation, estrogen levels in your body go down and your breast cells stop growing. In breast cancer, your breast cells make too many receptors, so when estrogen levels go up in your body, like during your normal menstrual cycle, your breast cells grow abnormally, which is one hallmark of cancer. If other changes occur in your breast cells to form a cancerous growth, these estrogen and progesterone receptors make the cancer cells grow uncontrollably.
Hormone receptor positive, also referred to as ER+, ER/PR+ breast cancer, is the most common type of breast cancer, accounting for 70-80% of breast cancers. They are diagnosed by a pathologist based on analysis of hormone receptor proteins present in cancer cells in a biopsy and in the tumor after surgical removal. This type of breast cancer, like most breast cancers, is first treated by surgery to remove the tumor. Depending on stage and grade, the ER/PR+ breast cancers should be analyzed by tumor genomic tests like Oncotype Dx or MammaPrint, which helps predict how likely the cancer is to recur (i.e. come back) and if chemotherapy is necessary for treatment.
Follow-up treatments include hormone therapies that block the activity of estrogen in the body, like the drug Tamoxifen, drugs that block estrogen production by the body, aromatase inhibitors like Letrozole, Arimidex, and Exemestane, or drugs that degrade estrogen receptor like Fulvestrant. Other ER+ breast cancer treatments include drugs that block the activity of proteins that drive cell growth (CDK inhibitors), including Ribociclib, Palbociclib, and Abemaciclib. These are typically used in combination with endocrine therapies like Tamoxifen/AIs/Fulvestrant to treat metastatic breast cancer, which has spread to other parts of the body. For women diagnosed with cancer who haven’t yet undergone menopause, medically induced menopause may be recommended. These treatments reduce the risk of the cancer from coming back, or recurring. They do come with some not-so-great side effects, which your oncologist should consider and help you with. Quality of life should always be a consideration when it comes to cancer treatment.
For more on hormone receptor positive breast cancer, check out the American Cancer Association. As with other subtypes of breast cancer, early detection increases your chance of survival, so keep up with your routine mammograms and self-exams.
The book has been out for about 3 weeks and I’ve been thrilled/nervous/pee-my-pants-excited to see my Amazon ranking as well as checking for ratings and reviews on Amazon, Goodreads, and other retail sites like Barnes & Noble, Walmart, Google Books, and Rowman & Littlefield!
For a brief, beautiful, shining moment, it was the #1 New Release in Breast Cancer and Oncology on Amazon, and I have the screenshots to commemorate it!
Pics or it didn’t happen!
Want a sneak peek? Of course you do! Here’s an excerpt from Chapter 16 that deals with an exciting new development in cancer research and treatment – harnessing the patient’s own immune system to seek out and destroy cancer cells through immune checkpoint inhibitors.
EXCERPT
I’ll also take comfort in the fact that we’re getting new weapons in the arsenal for fighting breast cancer. Antitumor immunity is the hottest thing to hit the field of cancer research since the 2001 approval of Gleevec (a game-changer drug used to treat chronic myelogenous leukemia that targets the oncoprotein product of the Philadelphia chromosome that drives the disease) and the 2006 approval of Gardasil (first vaccine targeting the human papilloma virus strains that cause most cervical cancers). Recently Frontiers in Immunology published the history of antitumor immunity efforts leading to the development of immune-checkpoint inhibitors available in the clinic today, the use of engineered T-cells taken from patients and altered to fight their cancer, and oncolytic viruses.2 I’ll go over the basics, including how antitumor immunity works and the challenges we still face in getting tumors to respond.
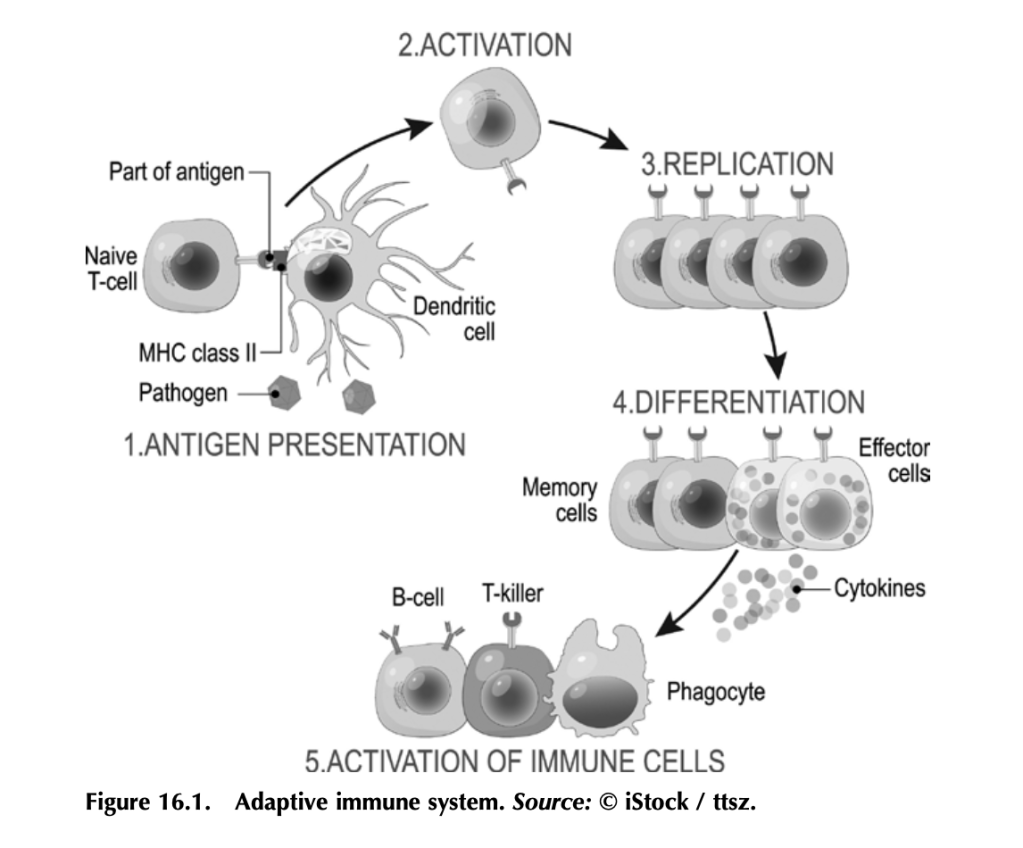
Before we get into how antitumor immunity works, we need to understand how the immune system works to fight infection. It’s a complex beast, but here are some basics. Your immune system functions to mount a rapid and robust defense when your body encounters a pathogen (e.g., a virus or bacteria that causes disease) in your daily life. The arm of the immune system that does this is called the adaptive immune system (figure 16.1). The other arm is the innate immune system, which includes natural barriers like skin, the tiny hairs and mucous in your nose, and stomach acid. The adaptive immune system is what antitumor immunity treatments harness. It is also altered by tumors to suppress tumor immune responses and exploited to work for the tumor. (More on that in a bit.)
The adaptive immune system works like this: Specialized cells identify a potential threat (e.g., an infection), and they carry information about that threat in the form of bits of protein called antigens to other immune cells. If the threat is credible, those immune cells get activated and fight the threat. First the specialized cells that identify a potential threat patrol your body, looking for something suspicious. Cells like macrophages and dendritic cells, which roam around various organs and tissues, find pathogens (a bacteria, virus, or other microbe that causes disease) or unhealthy cells infected by pathogens, and eat them (the fancy term is phagocytosis). Infected or damaged cells send out protein signals called cytokines as a distress call to attract these patrolling macrophages and dendritic cells. While “digesting” the bacteria or infected cell, macrophages and dendritic cells salvage proteins or pieces of proteins—antigens—that identify the bacteria or virus as “other,” and they present these to immune cells, usually in lymph nodes, which in turn mount an immune response. Macrophages and dendritic cells are known as professional antigen presenting cells (APCs).
When activated by APCs, immune cells called B-cells produce antibodies against the antigen, which can do a lot of things to fight an infection. Some antibodies neutralize the pathogen by binding it and stopping it from entering a cell. Other antibodies tag infected cells as a signal for other immune cells to come and kill them. Others coat pathogens or infected cells in a process called opsonization (meaning “the process of making tasty”), which signals other cells like macrophages to come and eat the coated pathogens or cells. Specialized B-cells called memory B-cells store the information about the antigen so your immune system can recognize the pathogen when it hits you again and mount a faster immune response.
Other immune cells called T-cells, which are particularly relevant to antitumor immunity, become activated by APCs and mount a different kind of immune response. Cytotoxic T-cells seek out and kill infected or damaged cells, and helper T-cells help activate B-cells so they make antibodies, activate cytotoxic T-cells, and activate macrophages to go eat nasty invaders and infected cells. Memory T-cells also store information about past infections to mount a rapid, strong response the next time your body sees it.
That’s a simplified but hopefully digestible explanation of immunity and the major players (there are other immune cells, but APCs, B-cells, and T-cells are the biggies).
Memory is key to protection, and memory is built by exposure to pathogens.
Put a pin in that concept for when we get to anticancer vaccines, and also remember what T-cells do for when we get to engineered CAR T-cells and oncolytic viruses.
Working out how to harness your body’s own immune system to fight cancer isn’t a new idea. It’s been under investigation since the nineteenth century. In fact, in chapter 5 we covered the way trastuzumab (trade name Herceptin), a humanized anti-HER2 antibody, targets HER2-expressing breast cancer cells for death. Herceptin and other monoclonal antibodies mimic the natural activity of antibody- producing B-cells to deliver therapies and tag cancer antigen–expressing cells for immune-mediated destruction. But it was the discovery of checkpoint inhibitors—proteins that put T-cells in a state of exhaustion and inactivity in pathways that are exploited by many cancers— that led to the first molecularly targeted therapies designed to boost antitumor immunity. Doctors James Allison and Tasuku Honjo pioneered this Nobel Prize–winning work.3
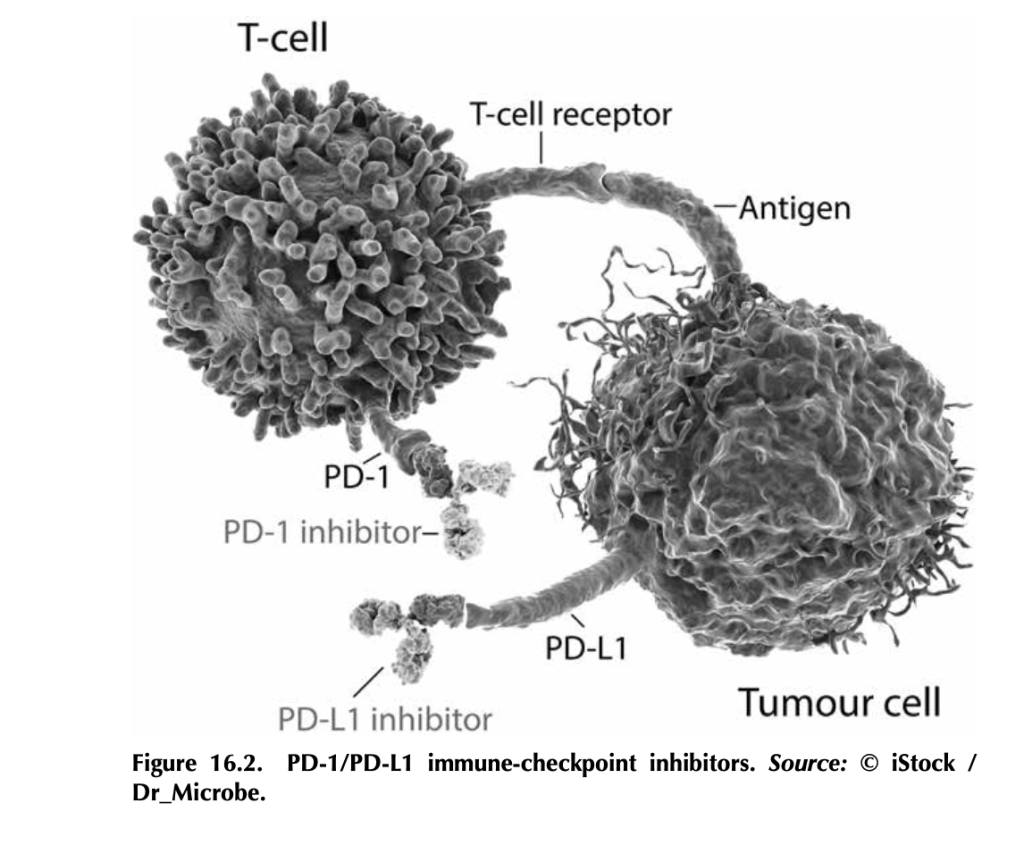
What are immune-checkpoint inhibitors, and how do they work? T- cells, particularly cytotoxic T-cells that actively kill their targets, bind to antigens on tumor cells through their T-cell receptors. But tumor cells, being the adaptable beasts that they are, can produce proteins like PD-L1 (programmed death ligand 1), which bind to PD-1 (programmed cell death protein 1), proteins on T-cells. This interaction tells the T- cell to stand down by tricking it into thinking that the tumor cell is “self” and should be protected. Signaling networks like this normally promote self-tolerance so that your immune system doesn’t attack your own healthy cells (figure 16.2). In tumors, it works by telling tumor- infiltrating T-cells, if present, to go into a state of inactivity. Drugs that target PD-L1—like atezolizumab (trade name Tecentriq), durvalumab (trade name Imfinzi), and avelumab (trade name Bavencio)—and drugs that target PD-1—like nivolumab (trade name Opdivo) and pembrozolimuab (trade name Keytruda)—are FDA-approved mono- clonal-antibody therapies that block interactions between PD-1/PD-L1 to unleash an antitumor immune response.4
Other immune-checkpoint molecules exploited by cancers include cytotoxic T lymphocyte antigen 4 (CTLA-4), the target of the first FDA-approved immune-checkpoint inhibitor ipilimumab (trade name Yervoy). Approved in 2011 for advanced melanoma, this drug had remarkable results. In fact, over 20 percent of the patients enrolled in the initial ipilimumab clinical trials (before the 2011 approval) are still alive and show no evidence of disease (NED).
There’s some incredible potential in targeting checkpoint inhibitors.
CTLA-4 is part of a cellular-signaling pathway that normally fine- tunes immune responses. CTLA-4 and a similar receptor, CD28, are expressed on two different T-cell types: (1) CD4+ helper T-cells, which help activate other immune cells to mediate adaptive immune responses, and (2) CD8+ cytotoxic T-cells, those cells that kill infected cells, damaged cells, and, if properly activated, tumor cells. Antigen- presenting cells make a protein called B7, which can bind to either CD28 or CTLA-4 on T-cells, and the effects on T-cell function are very different depending on what B7 binds. If it binds to CD28, B7 activates T-cell responses as a part of a complex of proteins that includes the T-cell receptor. Binding of B7 to CTLA-4 shuts down T- cell functions. CTLA-4 probably serves as protection from self-antigen recognition by inducing immune suppression, since laboratory mouse models engineered to not express CTLA-4 die from autoimmunity. This is the aspect of CTLA-4 function that gets highjacked by tumor cells. Drugs like ipilimumab block the suppressive activity of CTLA-4, which can allow T-cells to attack tumor cells.5
Here’s the kicker: The tumor actually has to have infiltrating T-cells for this to work, and not all tumors do. Tumors with T-cells that can be activated to fight the tumor are called “hot,” whereas tumors without T-cells are “cold.” One of the most aggressively researched topics in tumor immunology right now is how to make a cold tumor hot and thus responsive to antitumor immune therapies.
This is especially important for breast cancer, since most subtypes produce cold tumors. Right now, immune-checkpoint therapies are only approved for advanced triple-negative breast cancers that make the PD-L1 protein. Not all triple-negative breast cancers make PD-L1. Ongoing research is looking to expand the use of immune therapy in inflammatory breast cancer and the HER2+ subtype.6 Hopefully, with more research, we’ll figure out how to make more tumors responsive to immune therapy by making them hot (full of T-cells) and by discover- ing other immune checkpoints that can be targeted.
3. Heidi Ledford, Holly Else, and Matthew Warren, “Cancer Immunologists Scoop Medicine Nobel Prize,” Nature, October 1, 2018, https://www.nature. com/articles/d41586-018-06751-0.
4. See American Cancer Society medical and editorial content team, “Immunotherapy for Breast Cancer,” Treating Breast Cancer, American Cancer Society, Cancer.org, last revised December 3, 2020, https://www.cancer.org/ cancer/breast-cancer/treatment/immunotherapy.html.
6. Devon Carter, “Does Immunotherapy Treat Breast Cancer?” MD Anderson Center (website), University of Texas, March 26, 2021, https://www .mdanderson.org/cancerwise/does-immunotherapy-treat-breast-cancer.h00 -159385101.html.
Metastasis – the spread of cancer from its initial tissue of origin to another part (or parts) of the patient’s body – is deadly. Metastatic disease is, by and large, what kills people with cancer. It is an ongoing challenge for healthcare providers and researchers, and, as you may have guessed, it’s complicated.
But what exactly is metastasis? How does the process work? And why is it so hard to treat? I’ll cover what we know in this blog post, current and emerging therapies, and ongoing research designed to treat metastatic disease and allow cancer patients to survive and thrive by keeping their metastatic tumors at bay.
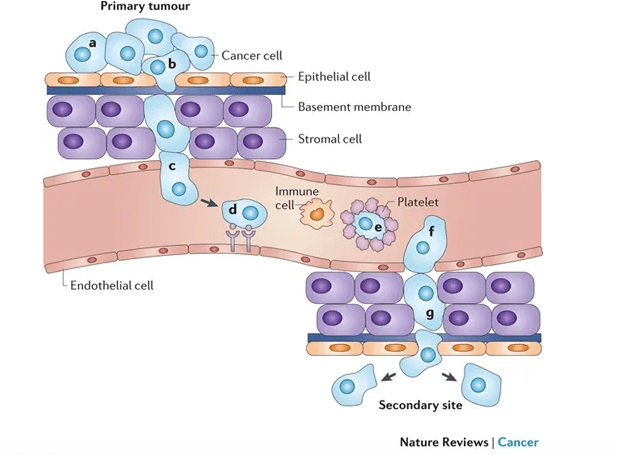
Here are the basics: tumor cells that have the ability to break away from the primary mass and invade the surrounding tissue can travel through the body via circulation (by entering the bloodstream directly or or by entering lymph nodes and from there, lymph vessels that shunt fluid back into circulation), invade a secondary organ, and begin to grow and form a new mass at the second location.
This isn’t easy for cancer cells to do. One of my grad school professors once referred to metastatic cancer cells as the decatheletes of cancer cells. Losing cell-cell contact with the tumor mass, invading the surrounding tissue, which is often a hostile environment without resources available to the primary tumor mass, is risky. Entering circulation is even more risky. The cells of origin for cancer cells are not normally equipped to withstand shear forces produced by flowing blood. They also have to avoid detection and destruction by immune cells, not only in circulation, but within the tissue of origin and within lymph nodes. Immune cells are programmed to seek out and destroy unhealthy cells, which may harbor bacterial or viral pathogens that threaten the body as a whole. Metastatic cells also have to crawl along blood vessel walls or hitch a ride on platelets, surviving in circulation without the resources available within the primary tumor mass.
From Schroeder et al. 2011 Nat Rev Can 12: 39-50.
If the metastatic cells manage to survive breaking away from the primary mass, evade vigilant immune cells, and travel through the harsh environment of the circulatory system, they face the arguably greater challenge of exiting circulation and setting up shop in an entirely different organ system that may or may not be similar to their original home. Think of them as colonists. They need to secure a space to live, gather resources from an unfamiliar landscape by competing with native cells that are better equipped because they belong, and they need to adapt and change the behavioral programs controlled by their genetic instructions in order to grow and establish a new tumor.
For breast cancer cells, common sites of metastasis include liver, lung, bone, and brain. Why those sites? One theory, the “seed and soil” hypothesis, argues that tumor cells are like plant seeds, which travel in all directions but can only live and grow if they land in compatible soil, meaning something about these particular organ environments allows tumor cells to take root. It’s an old theory, first posed by English surgeon Stephen Paget after studying autopsy records of 735 patients who died of breast cancer and spotting patterns.
During the process of invasion and metastatic spread, cancer cells experience a lot of pressures, and combined with a relatively unstable genome (covered in previous post), these pressures select for survival of cells that adapt in a process comparable to evolution by natural selection: cells that survive long enough to divide are more likely to pass favorable traits to their daughter cells. One effect of this process is that tumors formed by metastatic cells are often very different from the primary tumor, making them resistant to the therapies used to treat the primary tumor as well as other treatments. Often, they cannot be removed easily by surgery, are resistant to or quickly become resistant to chemotherapy, radiation, and targeted therapies, and grow at a rate that depletes the patient’s body of life-giving resources and causing the organs in which they are lodged to fail. In a nutshell, metastatic disease is incredibly difficult to treat.
So what can we do about it? The good news is that it is possible to manage metastatic disease in some cases, allowing patients to live longer with better quality of life. More therapies are extending the lives of patients living with metastatic breast cancer, including CDK inhibitors like Palbociclib [Ibrance; other similar drugs include Abemaciclib (Verzenio), palbociclib (Ibrance) and ribociclib (Kisqali)] that target cyclin dependent kinases that drive rapid proliferation of cancer cells, slowing their growth. Others include HER2 antibody-chemotherapy drug conjugates (delivers chemotherapy more specifically to HER2+ metastatic breast cancer cells), second-line HER2 targeted therapies, PI3-kinase inhibitors (which target a signaling pathway that is aberrantly activated in ~60% of cancers), PARP inhibitors (block DNA damage repair pathways to make cancer cells respond better to DNA damage inducing chemotherapy), and immune checkpoint inhibitors (activates T-cells in tumors and allows them to kill metastatic tumor cells) among others. See previous post for information about some of these molecular targets. For more on tumor immunology, click here.
These therapies extend the lives of metastatic breast cancer patients, but they are still a temporary fix. As mentioned above, metastatic tumor cells are tough, incredibly adaptable, and able to develop resistance to therapy. Another approach involves finding a way to induce or maintain tumor dormancy, a state in which tumor cells survive but remain quiescent rather than growing rapidly. Many metastatic lesions can persist in a state of dormancy for decades, and we do not yet understand what keeps them dormant, and perhaps more importantly, what activates their growth. But as researchers unravel the molecular mechanism that regulate dormancy and reactivation, new therapies can be developed to maintain dormancy – thus allowing cancer patients to survive and thrive during a normal lifespan in spite of their tumor burden.
Take home message: metastasis is a complex process that enables invading tumor cells to break away from the primary tumor, travel through the patient’s body, and set up shop in different organs. They are difficult to treat and are the main cause of cancer deaths, but current and emerging therapies to manage metastatic cancer are allowing patients to live longer, better quality lives.